Some Examples for Usage
This is not really an extensive usage guide but it is there to give you an idea about how to use different modules in the library together to get the most out of it. Here we are assuming that we have a project description that we are interested.
I’ve worked on NMD (nonsense mediated mRNA decay) for my thesis work and this pathway holds a special place in my heart. During my thesis work, I focused on trying to find the molecular differences between normal and premature termination using several different methods. So, for this tutorial let’s assume we are doing just that.
Project description
This will be our project description that we will be using.
Project Description
Nonsense-mediated mRNA decay (NMD) plays an important role in eukaryotic gene expression and mRNA degradataion through decapping and poly(A) shorteninig, yet the scope and the defining features of NMD-targeted transcripts remain elusive. This project aims to understand how NMD manages to differentiate between different substrates and which features of mRNAs are important for selection. Additionally we aim to understand the mechanisitic underpinnnigs of NMD as well as the factors involved in promoting mRNA degratdataion. We see to illustrate the differences between normal and premature termination both in terms of kinetic or other biochemical properties as well as any different factors involved in this differentiation if any. We plan to use in vitro, in vivo, ex vivo and in silico analysis whenever applicable to accomplish this goal
Literature search
Like any project we will start with a literature search and collect all the information that we will need to generate a set of valid hypotheses and a research plan.
We will first perform a pubmned search and then we will go over some of the papers and find ones that are relevant to our project and download and process them.
from benchmate.literature.literature import LitSearch, Paper, PaperInfo
litsearch=LitSearch()
results=litsearch.search("nonsense mediated mrna decay", database="pubmed", max_results=10_000)
len(results)
3042
We got 3000+ papers, it is unlikely that these are all relevant to our project. I will get some info about these papers and then decide if they are relevant with a few lines of code. In this example I’m not going to go through all of them since that will take a bit of time, but I will go over every 60th paper to speed things up a little bit.
import time
papers=[]
for i in range(len(results)):
if i%60==0:
p=Paper(paper_id=paper_id, id_type="pubmed")
p.get_abstract()
papers.append(p)
time.sleep(1) # I'm waiting 1 second between each query so I dont get kicked out of ncbi servers
else:
continue
Now I have collected abtracts, authors etc. for all the papers. I will then use my project description and the paper abstract to determine if these papers are relevant to what I’m interested in. Under the hood, I’m taking my project description and the paper abstract and semantically chunking them then these chunks are compared to each other and the max score for each column and row is calculated. Then I just take the average of this. Before we move on let’s take a look at one of the paper class instances. All the information that is related to the paper is under the info attribute.
p.info.abstract
'Premature termination codons (PTCs) can result in the production of truncated proteins or the degradation of messenger RNAs by nonsense-mediated mRNA decay (NMD). Which of these outcomes occurs can alter the effect of a mutation, with the engagement of NMD being dependent on a series of rules. Here, by applying these rules genome-wide to obtain a resource called NMDetective, we explore the impact of NMD on genetic disease and approaches to therapy. First, human genetic diseases differ in whether NMD typically aggravates or alleviates the effects of PTCs. Second, failure to trigger NMD is a cause of ineffective gene inactivation by CRISPR-Cas9 gene editing. Finally, NMD is a determinant of the efficacy of cancer immunotherapy, with only frameshifted transcripts that escape NMD predicting a response. These results demonstrate the importance of incorporating the rules of NMD into clinical decision-making. Moreover, they suggest that inhibiting NMD may be effective in enhancing cancer immunotherapy.'
p.info.title #and other things
'The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy.'
from benchmate.literature.paper_processor import PaperProcessor
from benchmate.config import *
The models I will be using for the demo are described under benchmate.config.py. A good amount of trial and error went into these decisions to keep things lightweight and accurate. While you can change these if you are using the modules independenty some of the features that are being developed have values hardcoded to them. I am aware of this limtation and will be resolving them once all the features are in place.
Let’s take a literature section of the config
literature
{'vl_model': {'model': {'name': 'Qwen/Qwen2.5-VL-3B-Instruct',
'config': {'cache_dir': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/hf_models'}},
'processor': {'name': 'Qwen/Qwen2.5-VL-3B-Instruct',
'config': {'cache_dir': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/hf_models'}},
'table_prompt': 'You are an expert biologist who is responsible for reading and interpreting scientific tables. For a given table from a scientific paper interpret the table. Do not provide comments on whether the table is well done or not. Do not provide extra text on describing that you are looking at table from a scientific publication. Give an overall conclusion about what the tables tells us.',
'figure_prompt': 'You are an expert biologist who is responsible for reading and interpreting scientific figures. For a given figure from a scientific paper interpret the figure. Do not provide comments on whether the figure is well done or not. Do not provide extra text on describing that you are looking at figure from a scientific publication. Whenever possible very briefly describe each sections of the figure and then give an overall conclusion about what the figure tells us. '},
'lp_model': {'model_path': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/lp_model/model_final.pth',
'config_path': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/lp_model/config.yaml'},
'text_embedding_model': {'name': 'Qwen/Qwen3-Embedding-0.6B',
'config': {'cache_folder': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/hf_models'}},
'image_embedding_model': {'model': {'name': 'vidore/colpali-v1.3',
'config': {'cache_dir': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/hf_models'}},
'processor': {'name': 'vidore/colpali-v1.3',
'config': {'cache_dir': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/hf_models'}}},
'chunker_model': {'model': '/home/alper/Documents/packages/ccm_benchmate/benchmate/models/m2v_model/',
'min_sentences': 1,
'return_type': 'texts',
'threshold': 'auto',
'chunk_size': 100}}
Now that we know what we are deling with we can go ahead and calucate relevances.
processor=PaperProcessor(literature)
project_description="""Nonsense-mediated mRNA decay (NMD) plays an important role in eukaryotic gene expression and mRNA degradataion through decapping and poly(A) shorteninig, yet the scope and the defining features of NMD-targeted transcripts remain elusive. This project aims to understand how NMD manages to differentiate between different substrates and which features of mRNAs are important for selection. Additionally we aim to understand the mechanisitic underpinnnigs of NMD as well as the factors involved in promoting mRNA degratdataion. We see to illustrate the differences between normal and premature termination both in terms of kinetic or other biochemical properties as well as any different factors involved in this differentiation if any. We plan to use in vitro, in vivo, ex vivo and in silico analysis whenever applicable to accomplish this goal"""
scores=processor.text_score(project_description, papers)
scores_df=pd.DataFrame({"paper_id":[p.info.id for p in papers],
"paper_title":[p.info.title for p in papers],
"paper_score":scores})
scores_df=scores_df.sort_values("paper_score", ascending=False)
scores_df
This looks good, we can see that the higher the score the more relevant the paper seems to be. From my experience scores >0.55 are generally a pretty safe bet. I will then take all those papers and I will see if they have a download link.
relevant_paper_ids=scores_df["paper_id"][scores_df["paper_score"]>0.55].tolist()
relevant_papers=[paper for paper in papers if paper.info.id in relevant_paper_ids]
for paper in tqdm(relevant_papers):
try:
paper.search_info()
if paper.info.download_link is not None:
paper.download("./papers") #Make sure this folder exisits otheriwise you will get an error
except Exception as e:
print(e)
Due to reasons beyong my control, I cannot just get all the papers. Some papers are behind paywalls and I do not have a pubmed api key from my institution that I can use. Additionally, some papers cannot be downloaded programatically even though they are open access, this is again because of 3rd part server settings.
After all this now we can process the papers this includes a few steps you can do all of them or one of.
- We will extract all the text from the pdfs
- We will chunk and embed the text as we have done with all the abstracts
- We will extract the figures and tables as images
- We will embed the figures and tables using a computer vision model
- We will generate figure and table captions
The main reason for 5 is because pdfs are quirky files. You can include a lot of informatio about different parts of the document or none at all. The document will still look the same. They can be under different tag or metadata fields and this is different (or can be different) for each pdf. There is really no way to figure that out. Additionally, sometimes (this is especially true for large figures and tables) their captions can be on a different page, we are processing (again due to pdf quirks) each pdf page by page and we might not have the associated caption on the page we are woking on.
While cumbersome, to get aroung all these limitations we are generating out figure captions from scratch. The VL model does a decent job for the most part and we have the actual caption from the extracted text anyway.
to_process=relevant_papers[0] #pubmed id 29891560 I cannot include the pdf because copyright but its open access
processed=processor.pipeline([to_process], extract=True, embed_text=False, embed_images=False, interpret_images=True, embed_iterpretations=False)
#lets take a look at the first figure
from PIL import Image
from IPython.display import display
display(processed.info.figures[0])

#and its caption
processed.info.figure_interpretation[0]
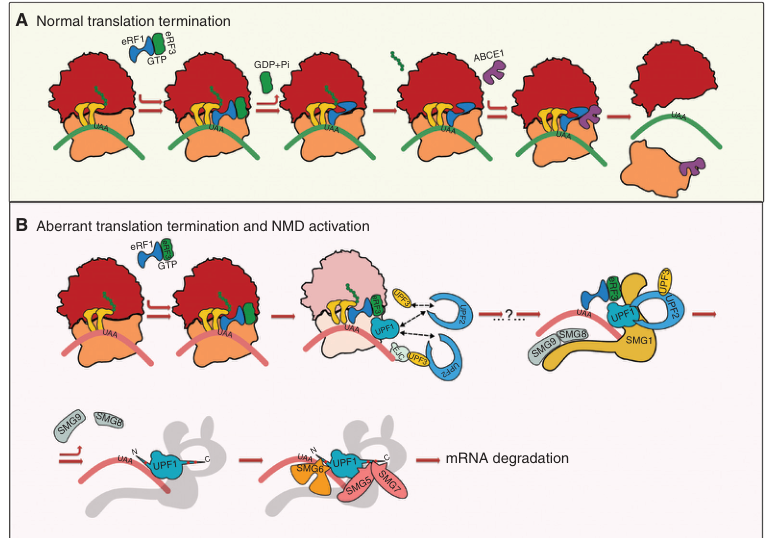
['**Figure A: Normal Translation Termination**\n\n- **Top Row:** The process begins with eEF1-GTP binding to the ribosome, which facilitates the release of the tRNA at the A site.\n- **Middle Row:** GDP+Pi is released, and the GTPase activity of eEF1 hydrolyzes GDP to GDP+Pi.\n- **Bottom Row:** ABCE1 (a protein involved in mRNA degradation) binds to the ribosome, marking it for degradation.\n\n**']
Not bad. If you want to get more information (and you should) there are some other methods you can run per paper.
processed.get_references() # will return a separate paper instance for each reference
processed.get_cited_by()
processed.get_related_works() #this is from openalex, I have nothing to do with it.
Coming Soon
All this processing is not for the fun of it. We are working on creating modules that will allow you to create searchable database that you can ask questions or perform keyword searches. We will update our package with these features very soon.
APIs module
Papers are one source of information, another sources is public databases, we have implemented numerous public databases within benchmate you can see our documentation for more details and how to use each of these. For this demo we will just use UniProt for a quick query
import pandas as pd
from benchmate.apis.uniprot import UniProt
uniprot=UniProt()
protein_results=uniprot.search("human nonsense mediated mrna decay, NMD", page_size=500)
protein_results=pd.DataFrame(protein_results)
protein_results
| id | gene | description | synonyms | organism | |
|---|---|---|---|---|---|
| 0 | Q9H0W8 | SMG9 | [Nonsense-mediated mRNA decay factor SMG9] | [C19orf61] | Homo sapiens |
| 1 | Q92540 | SMG7 | [Nonsense-mediated mRNA decay factor SMG7, [SM... | [C1orf16, EST1C, KIAA0250] | Homo sapiens |
| 2 | Q9UPR3 | SMG5 | [Nonsense-mediated mRNA decay factor SMG5, [ES... | [EST1B, KIAA1089] | Homo sapiens |
| 3 | Q86US8 | SMG6 | [Telomerase-binding protein EST1A, [Ever short... | [C17orf31, EST1A, KIAA0732] | Homo sapiens |
| 4 | Q8ND04 | SMG8 | [Nonsense-mediated mRNA decay factor SMG8, [Am... | [ABC2, C17orf71] | Homo sapiens |
| ... | ... | ... | ... | ... | ... |
| 659 | Q8IBN5 | None | [40S ribosomal protein S5, putative] | [] | Plasmodium falciparum (isolate 3D7) |
| 660 | A0AA88KHA3 | None | [60S ribosomal export protein NMD3] | [] | Naegleria lovaniensis |
| 661 | Q8IBJ9 | None | [Mago nashi protein homologue, putative] | [] | Plasmodium falciparum (isolate 3D7) |
| 662 | Q8IE18 | None | [RNA-binding protein, putative] | [] | Plasmodium falciparum (isolate 3D7) |
| 663 | A0A8I6A5Y3 | Ubbl1 | [Ubiquitin B like 2] | [Ubcl1] | Rattus norvegicus |
664 rows × 5 columns
I actually know what I’m looking for, I’m interested in the Upf1 gene and it’s protein product, let’s get it’s uniprot id and collect information about it.
goi=protein_results[protein_results["gene"]=="UPF1"]
goi
| id | gene | description | synonyms | organism | |
|---|---|---|---|---|---|
| 5 | Q92900 | UPF1 | [Regulator of nonsense transcripts 1, [ATP-dep... | [KIAA0221, RENT1] | Homo sapiens |
protein_info=uniprot.get_info("Q92900")
This will return a dizzying amount of information. But before we get to that, I want to show you the ApiCall dataclass. This is where all the information is stored and it also comes with a few neat features.
protein_info
ApiCall @ 2025-11-05 14:02:50.566128 with args:('Q92900',), kwargs:{}
You can see that this has a timestamp. Since information in databases get updated, if you want to go back and re-run this query you don’t have to repeat the whole process you can just do
protein_info.rerun()
ApiCall @ 2025-11-05 14:03:04.058575 with args:('Q92900',), kwargs:{}
You can see the different time stamps. Ok, let’s take a look at what we have returned
list(protein_info.results.keys()) # the api call results are stored under results.
['id',
'name',
'sequence',
'organism',
'gene',
'feature_types',
'comment_types',
'references',
'xref_types',
'xrefs',
'description',
'json',
'secondary_accessions',
'variation',
'interactions',
'mutagenesis',
'isoforms']
There is so much info here but some of these I would like to pay a bit more attention to one of them is xrefs these are the 3rd party databases that uniprot queries for you and you have their ids there. Some of these databases are implemented in benchmate as well and you can take these ids and run those queires.
The other one is the the references these are pubmed ids and we just looked at what you can do about those.
protein_info.results["xrefs"]
| type | id | properties | isoform | evidences | |
|---|---|---|---|---|---|
| 0 | EMBL | U65533 | {'molecule type': 'mRNA', 'protein sequence ID... | NaN | NaN |
| 1 | EMBL | U59323 | {'molecule type': 'mRNA', 'protein sequence ID... | NaN | NaN |
| 2 | EMBL | D86988 | {'molecule type': 'mRNA', 'protein sequence ID... | NaN | NaN |
| 3 | EMBL | AF074016 | {'molecule type': 'mRNA', 'protein sequence ID... | NaN | NaN |
| 4 | EMBL | AC003972 | {'molecule type': 'Genomic_DNA', 'protein sequ... | NaN | NaN |
| ... | ... | ... | ... | ... | ... |
| 169 | Pfam | PF04851 | {'match status': '1', 'entry name': 'ResIII'} | NaN | NaN |
| 170 | Pfam | PF18141 | {'match status': '1', 'entry name': 'UPF1_1B_d... | NaN | NaN |
| 171 | Pfam | PF09416 | {'match status': '1', 'entry name': 'UPF1_Zn_b... | NaN | NaN |
| 172 | SUPFAM | SSF52540 | {'match status': '1', 'entry name': 'P-loop co... | NaN | NaN |
| 173 | PROSITE | PS51997 | {'match status': '1', 'entry name': 'UPF1_CH_R... | NaN | NaN |
174 rows × 5 columns
Comments and features are interesting too, but they are not as organized, you can use get get_features, get_comments methods in the uniprot class to get different things.
protein_info.results["comment_types"]
{'ALTERNATIVE_PRODUCTS',
'CATALYTIC_ACTIVITY',
'DOMAIN',
'FUNCTION',
'INTERACTION',
'PTM',
'SEQUENCE_CAUTION',
'SIMILARITY',
'SUBCELLULAR_LOCATION',
'SUBUNIT',
'TISSUE_SPECIFICITY'}
#you will need to pass and api call specific to uniprot to this
uniprot.get_comments(protein_info.results, "FUNCTION")
[{'type': 'FUNCTION',
'text': [{'value': "RNA-dependent helicase required for nonsense-mediated decay (NMD) of aberrant mRNAs containing premature stop codons and modulates the expression level of normal mRNAs (PubMed:11163187, PubMed:16086026, PubMed:18172165, PubMed:21145460, PubMed:21419344, PubMed:24726324). Is recruited to mRNAs upon translation termination and undergoes a cycle of phosphorylation and dephosphorylation; its phosphorylation appears to be a key step in NMD (PubMed:11544179, PubMed:25220460). Recruited by release factors to stalled ribosomes together with the SMG1C protein kinase complex to form the transient SURF (SMG1-UPF1-eRF1-eRF3) complex (PubMed:19417104). In EJC-dependent NMD, the SURF complex associates with the exon junction complex (EJC) (located 50-55 or more nucleotides downstream from the termination codon) through UPF2 and allows the formation of an UPF1-UPF2-UPF3 surveillance complex which is believed to activate NMD (PubMed:21419344). Phosphorylated UPF1 is recognized by EST1B/SMG5, SMG6 and SMG7 which are thought to provide a link to the mRNA degradation machinery involving exonucleolytic and endonucleolytic pathways, and to serve as adapters to protein phosphatase 2A (PP2A), thereby triggering UPF1 dephosphorylation and allowing the recycling of NMD factors (PubMed:12554878). UPF1 can also activate NMD without UPF2 or UPF3, and in the absence of the NMD-enhancing downstream EJC indicative for alternative NMD pathways (PubMed:18447585). Plays a role in replication-dependent histone mRNA degradation at the end of phase S; the function is independent of UPF2 (PubMed:16086026, PubMed:18172165). For the recognition of premature termination codons (PTC) and initiation of NMD a competitive interaction between UPF1 and PABPC1 with the ribosome-bound release factors is proposed (PubMed:18447585, PubMed:25220460). The ATPase activity of UPF1 is required for disassembly of mRNPs undergoing NMD (PubMed:21145460). Together with UPF2 and dependent on TDRD6, mediates the degradation of mRNA harboring long 3'UTR by inducing the NMD machinery (By similarity). Also capable of unwinding double-stranded DNA and translocating on single-stranded DNA (PubMed:30218034)",
'evidences': [{'code': 'ECO:0000250',
'source': {'name': 'UniProtKB',
'id': 'Q9EPU0',
'url': 'https://www.uniprot.org/uniprot/Q9EPU0'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '11163187',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/11163187',
'alternativeUrl': 'https://europepmc.org/abstract/MED/11163187'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '11544179',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/11544179',
'alternativeUrl': 'https://europepmc.org/abstract/MED/11544179'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '12554878',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/12554878',
'alternativeUrl': 'https://europepmc.org/abstract/MED/12554878'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '16086026',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/16086026',
'alternativeUrl': 'https://europepmc.org/abstract/MED/16086026'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '18172165',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/18172165',
'alternativeUrl': 'https://europepmc.org/abstract/MED/18172165'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '18447585',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/18447585',
'alternativeUrl': 'https://europepmc.org/abstract/MED/18447585'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '19417104',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/19417104',
'alternativeUrl': 'https://europepmc.org/abstract/MED/19417104'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '21145460',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/21145460',
'alternativeUrl': 'https://europepmc.org/abstract/MED/21145460'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '21419344',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/21419344',
'alternativeUrl': 'https://europepmc.org/abstract/MED/21419344'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '24726324',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/24726324',
'alternativeUrl': 'https://europepmc.org/abstract/MED/24726324'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '25220460',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/25220460',
'alternativeUrl': 'https://europepmc.org/abstract/MED/25220460'}},
{'code': 'ECO:0000269',
'source': {'name': 'PubMed',
'id': '30218034',
'url': 'http://www.ncbi.nlm.nih.gov/pubmed/30218034',
'alternativeUrl': 'https://europepmc.org/abstract/MED/30218034'}}]}]}]
Well, this is a protein, where is its sequence?
protein_info.results["sequence"]
'MSVEAYGPSSQTLTFLDTEEAELLGADTQGSEFEFTDFTLPSQTQTPPGGPGGPGGGGAGGPGGAGAGAAAGQLDAQVGPEGILQNGAVDDSVAKTSQLLAELNFEEDEEDTYYTKDLPIHACSYCGIHDPACVVYCNTSKKWFCNGRGNTSGSHIVNHLVRAKCKEVTLHKDGPLGETVLECYNCGCRNVFLLGFIPAKADSVVVLLCRQPCASQSSLKDINWDSSQWQPLIQDRCFLSWLVKIPSEQEQLRARQITAQQINKLEELWKENPSATLEDLEKPGVDEEPQHVLLRYEDAYQYQNIFGPLVKLEADYDKKLKESQTQDNITVRWDLGLNKKRIAYFTLPKTDSGNEDLVIIWLRDMRLMQGDEICLRYKGDLAPLWKGIGHVIKVPDNYGDEIAIELRSSVGAPVEVTHNFQVDFVWKSTSFDRMQSALKTFAVDETSVSGYIYHKLLGHEVEDVIIKCQLPKRFTAQGLPDLNHSQVYAVKTVLQRPLSLIQGPPGTGKTVTSATIVYHLARQGNGPVLVCAPSNIAVDQLTEKIHQTGLKVVRLCAKSREAIDSPVSFLALHNQIRNMDSMPELQKLQQLKDETGELSSADEKRYRALKRTAERELLMNADVICCTCVGAGDPRLAKMQFRSILIDESTQATEPECMVPVVLGAKQLILVGDHCQLGPVVMCKKAAKAGLSQSLFERLVVLGIRPIRLQVQYRMHPALSAFPSNIFYEGSLQNGVTAADRVKKGFDFQWPQPDKPMFFYVTQGQEEIASSGTSYLNRTEAANVEKITTKLLKAGAKPDQIGIITPYEGQRSYLVQYMQFSGSLHTKLYQEVEIASVDAFQGREKDFIILSCVRANEHQGIGFLNDPRRLNVALTRARYGVIIVGNPKALSKQPLWNHLLNYYKEQKVLVEGPLNNLRESLMQFSKPRKLVNTINPGARFMTTAMYDAREAIIPGSVYDRSSQGRPSSMYFQTHDQIGMISAGPSHVAAMNIPIPFNLVMPPMPPPGYFGQANGPAAGRGTPKGKTGRGGRQKNRFGLPGPSQTNLPNSQASQDVASQPFSQGALTQGYISMSQPSQMSQPGLSQPELSQDSYLGDEFKSQIDVALSQDSTYQGERAYQHGGVTGLSQY'
Sequence modulde
I think this provides a good segue to the sequence module, this module has 2 main classes, Sequence and SequenceList
from benchmate.sequence.sequence import Sequence, SequenceList
myseq=Sequence(name="upf1", sequence=protein_info.results["sequence"])
Sequence objects have quite a few different methods associated with them, you can see what they are in the documentation, here I want to demonostrate the SequenceList so I will generate a few more sequences by generateing some version of the same sequence.
myseq2=myseq.mutate(0, "T")
myseq3=myseq.delete(10, 35)
seqlist=SequenceList([myseq, myseq2, myseq3])
seqlist.ClustalOmega()
(['MSVEAYGPSSQTLTFLDTEEAELLGADTQGSEFEFTDFTLPSQTQTPPGGPGGPGGGGAGGPGGAGAGAAAGQLDAQVGPEGILQNGAVDDSVAKTSQLLAELNFEEDEEDTYYTKDLPIHACSYCGIHDPACVVYCNTSKKWFCNGRGNTSGSHIVNHLVRAKCKEVTLHKDGPLGETVLECYNCGCRNVFLLGFIPAKADSVVVLLCRQPCASQSSLKDINWDSSQWQPLIQDRCFLSWLVKIPSEQEQLRARQITAQQINKLEELWKENPSATLEDLEKPGVDEEPQHVLLRYEDAYQYQNIFGPLVKLEADYDKKLKESQTQDNITVRWDLGLNKKRIAYFTLPKTDSGNEDLVIIWLRDMRLMQGDEICLRYKGDLAPLWKGIGHVIKVPDNYGDEIAIELRSSVGAPVEVTHNFQVDFVWKSTSFDRMQSALKTFAVDETSVSGYIYHKLLGHEVEDVIIKCQLPKRFTAQGLPDLNHSQVYAVKTVLQRPLSLIQGPPGTGKTVTSATIVYHLARQGNGPVLVCAPSNIAVDQLTEKIHQTGLKVVRLCAKSREAIDSPVSFLALHNQIRNMDSMPELQKLQQLKDETGELSSADEKRYRALKRTAERELLMNADVICCTCVGAGDPRLAKMQFRSILIDESTQATEPECMVPVVLGAKQLILVGDHCQLGPVVMCKKAAKAGLSQSLFERLVVLGIRPIRLQVQYRMHPALSAFPSNIFYEGSLQNGVTAADRVKKGFDFQWPQPDKPMFFYVTQGQEEIASSGTSYLNRTEAANVEKITTKLLKAGAKPDQIGIITPYEGQRSYLVQYMQFSGSLHTKLYQEVEIASVDAFQGREKDFIILSCVRANEHQGIGFLNDPRRLNVALTRARYGVIIVGNPKALSKQPLWNHLLNYYKEQKVLVEGPLNNLRESLMQFSKPRKLVNTINPGARFMTTAMYDAREAIIPGSVYDRSSQGRPSSMYFQTHDQIGMISAGPSHVAAMNIPIPFNLVMPPMPPPGYFGQANGPAAGRGTPKGKTGRGGRQKNRFGLPGPSQTNLPNSQASQDVASQPFSQGALTQGYISMSQPSQMSQPGLSQPELSQDSYLGDEFKSQIDVALSQDSTYQGERAYQHGGVTGLSQY',
'TSVEAYGPSSQTLTFLDTEEAELLGADTQGSEFEFTDFTLPSQTQTPPGGPGGPGGGGAGGPGGAGAGAAAGQLDAQVGPEGILQNGAVDDSVAKTSQLLAELNFEEDEEDTYYTKDLPIHACSYCGIHDPACVVYCNTSKKWFCNGRGNTSGSHIVNHLVRAKCKEVTLHKDGPLGETVLECYNCGCRNVFLLGFIPAKADSVVVLLCRQPCASQSSLKDINWDSSQWQPLIQDRCFLSWLVKIPSEQEQLRARQITAQQINKLEELWKENPSATLEDLEKPGVDEEPQHVLLRYEDAYQYQNIFGPLVKLEADYDKKLKESQTQDNITVRWDLGLNKKRIAYFTLPKTDSGNEDLVIIWLRDMRLMQGDEICLRYKGDLAPLWKGIGHVIKVPDNYGDEIAIELRSSVGAPVEVTHNFQVDFVWKSTSFDRMQSALKTFAVDETSVSGYIYHKLLGHEVEDVIIKCQLPKRFTAQGLPDLNHSQVYAVKTVLQRPLSLIQGPPGTGKTVTSATIVYHLARQGNGPVLVCAPSNIAVDQLTEKIHQTGLKVVRLCAKSREAIDSPVSFLALHNQIRNMDSMPELQKLQQLKDETGELSSADEKRYRALKRTAERELLMNADVICCTCVGAGDPRLAKMQFRSILIDESTQATEPECMVPVVLGAKQLILVGDHCQLGPVVMCKKAAKAGLSQSLFERLVVLGIRPIRLQVQYRMHPALSAFPSNIFYEGSLQNGVTAADRVKKGFDFQWPQPDKPMFFYVTQGQEEIASSGTSYLNRTEAANVEKITTKLLKAGAKPDQIGIITPYEGQRSYLVQYMQFSGSLHTKLYQEVEIASVDAFQGREKDFIILSCVRANEHQGIGFLNDPRRLNVALTRARYGVIIVGNPKALSKQPLWNHLLNYYKEQKVLVEGPLNNLRESLMQFSKPRKLVNTINPGARFMTTAMYDAREAIIPGSVYDRSSQGRPSSMYFQTHDQIGMISAGPSHVAAMNIPIPFNLVMPPMPPPGYFGQANGPAAGRGTPKGKTGRGGRQKNRFGLPGPSQTNLPNSQASQDVASQPFSQGALTQGYISMSQPSQMSQPGLSQPELSQDSYLGDEFKSQIDVALSQDSTYQGERAYQHGGVTGLSQY',
'MSVEAYGPS-------------------------STDFTLPSQTQTPPGGPGGPGGGGAGGPGGAGAGAAAGQLDAQVGPEGILQNGAVDDSVAKTSQLLAELNFEEDEEDTYYTKDLPIHACSYCGIHDPACVVYCNTSKKWFCNGRGNTSGSHIVNHLVRAKCKEVTLHKDGPLGETVLECYNCGCRNVFLLGFIPAKADSVVVLLCRQPCASQSSLKDINWDSSQWQPLIQDRCFLSWLVKIPSEQEQLRARQITAQQINKLEELWKENPSATLEDLEKPGVDEEPQHVLLRYEDAYQYQNIFGPLVKLEADYDKKLKESQTQDNITVRWDLGLNKKRIAYFTLPKTDSGNEDLVIIWLRDMRLMQGDEICLRYKGDLAPLWKGIGHVIKVPDNYGDEIAIELRSSVGAPVEVTHNFQVDFVWKSTSFDRMQSALKTFAVDETSVSGYIYHKLLGHEVEDVIIKCQLPKRFTAQGLPDLNHSQVYAVKTVLQRPLSLIQGPPGTGKTVTSATIVYHLARQGNGPVLVCAPSNIAVDQLTEKIHQTGLKVVRLCAKSREAIDSPVSFLALHNQIRNMDSMPELQKLQQLKDETGELSSADEKRYRALKRTAERELLMNADVICCTCVGAGDPRLAKMQFRSILIDESTQATEPECMVPVVLGAKQLILVGDHCQLGPVVMCKKAAKAGLSQSLFERLVVLGIRPIRLQVQYRMHPALSAFPSNIFYEGSLQNGVTAADRVKKGFDFQWPQPDKPMFFYVTQGQEEIASSGTSYLNRTEAANVEKITTKLLKAGAKPDQIGIITPYEGQRSYLVQYMQFSGSLHTKLYQEVEIASVDAFQGREKDFIILSCVRANEHQGIGFLNDPRRLNVALTRARYGVIIVGNPKALSKQPLWNHLLNYYKEQKVLVEGPLNNLRESLMQFSKPRKLVNTINPGARFMTTAMYDAREAIIPGSVYDRSSQGRPSSMYFQTHDQIGMISAGPSHVAAMNIPIPFNLVMPPMPPPGYFGQANGPAAGRGTPKGKTGRGGRQKNRFGLPGPSQTNLPNSQASQDVASQPFSQGALTQGYISMSQPSQMSQPGLSQPELSQDSYLGDEFKSQIDVALSQDSTYQGERAYQHGGVTGLSQY'],
array([[0. , 0.000886, 0.009058],
[0.000886, 0. , 0.009058],
[0.009058, 0.009058, 0. ]]),
'((0:0.0004428699903655797,1:0.0004428699903655797):0.004086120054125786,2:0.004528990015387535):0.0;')
Here I have created a sequence list instances and I have calcualted pairwise sequence alignment. You can read from a fasta (and write to it), if you have more than 1 sequence in your fasta you will get a SequenceList otherwise you will get a Sequence.
Our protein upf1 doesn’t just have sequences, it also has structures, let’s see if we can find any structures in the protein_results from uniprot.
protein_info.results["xrefs"]["type"].drop_duplicates().tolist()
['EMBL',
'CCDS',
'RefSeq',
'PDB',
'PDBsum',
'AlphaFoldDB',
'EMDB',
'SMR',
'BioGRID',
'CORUM',
'DIP',
'FunCoup',
'IntAct',
'MINT',
'STRING',
'GlyCosmos',
'GlyGen',
'iPTMnet',
'MetOSite',
'PhosphoSitePlus',
'SwissPalm',
'BioMuta',
'DMDM',
'jPOST',
'MassIVE',
'PaxDb',
'PeptideAtlas',
'ProteomicsDB',
'Pumba',
'Antibodypedia',
'DNASU',
'Ensembl',
'GeneID',
'KEGG',
'MANE-Select',
'UCSC',
'AGR',
'CTD',
'DisGeNET',
'GeneCards',
'HGNC',
'HPA',
'MalaCards',
'MIM',
'neXtProt',
'OpenTargets',
'PharmGKB',
'VEuPathDB',
'eggNOG',
'GeneTree',
'HOGENOM',
'InParanoid',
'OMA',
'OrthoDB',
'PAN-GO',
'PhylomeDB',
'TreeFam',
'BRENDA',
'PathwayCommons',
'Reactome',
'SignaLink',
'SIGNOR',
'BioGRID-ORCS',
'CD-CODE',
'ChiTaRS',
'EvolutionaryTrace',
'GeneWiki',
'GenomeRNAi',
'Pharos',
'PRO',
'Proteomes',
'RNAct',
'Bgee',
'ExpressionAtlas',
'GO',
'CDD',
'FunFam',
'Gene3D',
'IDEAL',
'InterPro',
'PANTHER',
'Pfam',
'SUPFAM',
'PROSITE']
I see “PDB” in the list of cross references, let’s take a look.
protein_info.results["xrefs"][protein_info.results["xrefs"]["type"]=="PDB"]
| type | id | properties | isoform | evidences | |
|---|---|---|---|---|---|
| 10 | PDB | 2GJK | {'method': 'X-ray', 'chains': 'A=295-925', 're... | NaN | NaN |
| 11 | PDB | 2GK6 | {'method': 'X-ray', 'chains': 'A/B=295-925', '... | NaN | NaN |
| 12 | PDB | 2GK7 | {'method': 'X-ray', 'chains': 'A=295-925', 're... | NaN | NaN |
| 13 | PDB | 2IYK | {'method': 'X-ray', 'chains': 'A/B=115-272', '... | NaN | NaN |
| 14 | PDB | 2WJV | {'method': 'X-ray', 'chains': 'A/B=115-925', '... | NaN | NaN |
| 15 | PDB | 2WJY | {'method': 'X-ray', 'chains': 'A=115-925', 're... | NaN | NaN |
| 16 | PDB | 2XZO | {'method': 'X-ray', 'chains': 'A=295-925', 're... | NaN | NaN |
| 17 | PDB | 2XZP | {'method': 'X-ray', 'chains': 'A=295-925', 're... | NaN | NaN |
| 18 | PDB | 6EJ5 | {'method': 'X-ray', 'chains': 'A=295-925', 're... | NaN | NaN |
| 19 | PDB | 6Z3R | {'method': 'EM', 'chains': 'E=1085-1095', 'res... | NaN | NaN |
| 20 | PDB | 8RXB | {'method': 'X-ray', 'chains': 'A/D/E/I/L/P=115... | NaN | NaN |
Oh, a lot of strucures, I see one with lot’s of chains the last one, let’s get that one.
from benchmate.structure.structure import Structure
my_str=Structure(name="upf1", source="PDB", id="8RXB", file=None)
If you have a local pdb file you can used that under file otherwise you can (like we did here) specify an id from pdb or alphafolddb to download it. We saw that the structure had a lot of chains, let’s see them.
my_str.info.chains
array(['E', 'F', 'A', 'B', 'D', 'G', 'I', 'J', 'L', 'N', 'P', 'Q', 'E',
'A', 'D', 'I', 'L', 'P'], dtype='<U4')
There a many methods associated with structures, a quick example here, I’m going to calculate the contact between chains E and F
contacts=my_str.contacts("E", "F")
contacts[0:10]
[{'E': 818, 'F': 50, 'distance': 4.976994},
{'E': 885, 'F': 55, 'distance': 4.9693766},
{'E': 886, 'F': 94, 'distance': 4.873773},
{'E': 886, 'F': 99, 'distance': 4.001063},
{'E': 886, 'F': 101, 'distance': 4.5759163},
{'E': 886, 'F': 103, 'distance': 4.421472},
{'E': 887, 'F': 99, 'distance': 4.924966},
{'E': 887, 'F': 101, 'distance': 4.9591007},
{'E': 890, 'F': 90, 'distance': 4.9222174},
{'E': 890, 'F': 94, 'distance': 4.6336584}]
We can see the first 10 atoms and the distances between them. You can also do some fancy indexing if you want to select things.
my_str["A"][10:30]
array([
Atom(np.array([30.226, 17.509, 9.708], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="O", element="O"),
Atom(np.array([28.546, 19.227, 8.242], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="CB", element="C"),
Atom(np.array([27.528, 18.246, 7.703], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="CG", element="C"),
Atom(np.array([26.726, 17.717, 8.501], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="OD1", element="O"),
Atom(np.array([27.537, 18.004, 6.476], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="OD2", element="O"),
Atom(np.array([29.917, 20.715, 9.766], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="H", element="H"),
Atom(np.array([27.671, 19.234, 10.103], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="HA", element="H"),
Atom(np.array([28.318, 20.109, 7.908], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="HB2", element="H"),
Atom(np.array([29.419, 18.963, 7.914], dtype=float32), chain_id="A", res_id=3, ins_code="", res_name="ASP", hetero=False, atom_name="HB3", element="H"),
Atom(np.array([28.983, 17.7 , 11.581], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="N", element="N"),
Atom(np.array([29.668, 16.577, 12.209], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="CA", element="C"),
Atom(np.array([29.111, 15.239, 11.724], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="C", element="C"),
Atom(np.array([27.944, 15.141, 11.328], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="O", element="O"),
Atom(np.array([29.565, 16.629, 13.73 ], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="CB", element="C"),
Atom(np.array([30.48 , 17.621, 14.445], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="CG", element="C"),
Atom(np.array([30.222, 17.602, 15.952], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="CD1", element="C"),
Atom(np.array([31.938, 17.301, 14.126], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="CD2", element="C"),
Atom(np.array([28.364, 18.064, 12.053], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="H", element="H"),
Atom(np.array([30.606, 16.632, 11.966], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="HA", element="H"),
Atom(np.array([28.653, 16.866, 13.961], dtype=float32), chain_id="A", res_id=4, ins_code="", res_name="LEU", hetero=False, atom_name="HB2", element="H")
])
While we are on the topic of NMD, I want to see if I can get some small molecules related to NMD.
from benchmate.apis.ncbi import Ncbi
ncbi=Ncbi(email="alper.celik@sickkids.ca")
I’m going to search pubchem but before that I want to show you all the things you can search.
ncbi.databases
['pubmed',
'protein',
'nuccore',
'ipg',
'nucleotide',
'structure',
'genome',
'annotinfo',
'assembly',
'bioproject',
'biosample',
'blastdbinfo',
'books',
'cdd',
'clinvar',
'gap',
'gapplus',
'grasp',
'dbvar',
'gene',
'gds',
'geoprofiles',
'medgen',
'mesh',
'nlmcatalog',
'omim',
'orgtrack',
'pmc',
'proteinclusters',
'pcassay',
'protfam',
'pccompound',
'pcsubstance',
'seqannot',
'snp',
'sra',
'taxonomy',
'biocollections',
'gtr']
#I'm going search for something generic
pubchem_search=ncbi.search("pccompound", "ribosome", retmax=10000)
len(pubchem_search.results)
2
Oof, only 2! I’ll get both I guess
mols=ncbi.summary("pccompound", pubchem_search.results)
mols.results
[{'Item': [], 'Id': '168475931', 'CID': IntegerElement(168475931, attributes={}), 'SourceNameList': [], 'SourceIDList': [], 'SourceCategoryList': ['Chemical Vendors'], 'CreateDate': '2023/09/05 00:00', 'SynonymList': ['SEQ-9', 'GLXC-27148', 'EX-A13126', '23S bacterial ribosome inhibitor SEQ-9', 'HY-153222', 'CS-0655585'], 'MeSHHeadingList': [], 'MeSHTermList': [], 'PharmActionList': [], 'CommentList': [], 'IUPACName': '[(2S,3R,4R,5R,7S,9S,10S,11R,12S,13R)-7-[[1-(benzenesulfonamido)-2-methylpropan-2-yl]carbamoyloxy]-2-[(1R)-1-[(2S,3R,4R,5R,6R)-5-hydroxy-3,4-dimethoxy-6-methyloxan-2-yl]oxyethyl]-10-[(2S,3R,4Z,6R)-3-hydroxy-4-methoxyimino-6-methyloxan-2-yl]oxy-3,5,7,9,11,13-hexamethyl-6,14-dioxo-12-[[(2S,5R,7R)-2,4,5-trimethyl-1,4-oxazepan-7-yl]oxy]-oxacyclotetradec-4-yl] 3-methylbutanoate', 'CanonicalSmiles': 'CC1CC(OC(CN1C)C)OC2C(C(C(CC(C(=O)C(C(C(C(OC(=O)C2C)C(C)OC3C(C(C(C(O3)C)O)OC)OC)C)OC(=O)CC(C)C)C)(C)OC(=O)NC(C)(C)CNS(=O)(=O)C4=CC=CC=C4)C)OC5C(C(=NOC)CC(O5)C)O)C', 'IsomericSmiles': 'C[C@@H]1C[C@@H](O[C@H](CN1C)C)O[C@H]2[C@@H]([C@H]([C@H](C[C@](C(=O)[C@@H]([C@@H]([C@H]([C@H](OC(=O)[C@@H]2C)[C@@H](C)O[C@H]3[C@@H]([C@@H]([C@@H]([C@H](O3)C)O)OC)OC)C)OC(=O)CC(C)C)C)(C)OC(=O)NC(C)(C)CNS(=O)(=O)C4=CC=CC=C4)C)O[C@H]5[C@@H](/C(=N\\OC)/C[C@H](O5)C)O)C', 'RotatableBondCount': IntegerElement(21, attributes={}), 'MolecularFormula': 'C60H100N4O20S', 'MolecularWeight': '1229.500', 'TotalFormalCharge': IntegerElement(0, attributes={}), 'XLogP': '6.5', 'HydrogenBondDonorCount': IntegerElement(4, attributes={}), 'HydrogenBondAcceptorCount': IntegerElement(23, attributes={}), 'Complexity': '2300.000', 'HeavyAtomCount': IntegerElement(85, attributes={}), 'AtomChiralCount': IntegerElement(22, attributes={}), 'AtomChiralDefCount': IntegerElement(22, attributes={}), 'AtomChiralUndefCount': IntegerElement(0, attributes={}), 'BondChiralCount': IntegerElement(1, attributes={}), 'BondChiralDefCount': IntegerElement(1, attributes={}), 'BondChiralUndefCount': IntegerElement(0, attributes={}), 'IsotopeAtomCount': IntegerElement(0, attributes={}), 'CovalentUnitCount': IntegerElement(1, attributes={}), 'TautomerCount': NoneElement(attributes={}), 'SubstanceIDList': [], 'TPSA': '302', 'AssaySourceNameList': [], 'MinAC': '', 'MaxAC': '', 'MinTC': '', 'MaxTC': '', 'ActiveAidCount': IntegerElement(0, attributes={}), 'InactiveAidCount': NoneElement(attributes={}), 'TotalAidCount': IntegerElement(0, attributes={}), 'InChIKey': 'JCUPFQUUARRICQ-DXXMFXFBSA-N', 'InChI': 'InChI=1S/C60H100N4O20S/c1-31(2)25-44(65)80-49-37(8)51(41(12)79-57-53(74-18)52(73-17)46(66)40(11)78-57)82-55(69)39(10)50(81-45-26-33(4)64(16)29-35(6)76-45)36(7)48(83-56-47(67)43(63-75-19)27-34(5)77-56)32(3)28-60(15,54(68)38(49)9)84-58(70)62-59(13,14)30-61-85(71,72)42-23-21-20-22-24-42/h20-24,31-41,45-53,56-57,61,66-67H,25-30H2,1-19H3,(H,62,70)/b63-43-/t32-,33+,34+,35-,36+,37+,38+,39+,40+,41+,45-,46+,47+,48-,49+,50-,51-,52+,53+,56-,57-,60-/m0/s1'}, {'Item': [], 'Id': '6438338', 'CID': IntegerElement(6438338, attributes={}), 'SourceNameList': [], 'SourceIDList': [], 'SourceCategoryList': ['Chemical Vendors', 'Legacy Depositors', 'Subscription Services', 'Curation Efforts', 'Research and Development', 'Governmental Organizations'], 'CreateDate': '2006/04/28 00:00', 'SynonymList': ['volkensin', 'Volkensin (protein)', '91933-11-8', 'RIP protein, A volkensii Harms', 'Ribosome-inactivating protein type 2, Adenia volkensii', '((1R,2R,4R,6S,8R,11R,12S,13R,16R,17R,19S,20R)-17-acetyloxy-8-(furan-3-yl)-4,12-dihydroxy-1,9,11,16-tetramethyl-5,14-dioxapentacyclo(11.6.1.02,11.06,10.016,20)icos-9-en-19-yl) (E)-2-methylbut-2-enoate', '[(1R,2R,4R,6S,8R,11R,12S,13R,16R,17R,19S,20R)-17-acetyloxy-8-(furan-3-yl)-4,12-dihydroxy-1,9,11,16-tetramethyl-5,14-dioxapentacyclo[11.6.1.02,11.06,10.016,20]icos-9-en-19-yl] (E)-2-methylbut-2-enoate', 'RefChem:194620', 'DTXSID201058638', 'Adenia volkesii toxin', 'Protein volkensin', 'Volkensin (glycoprotein)', 'CHEMBL4872878', 'SCHEMBL29359064', '1416549-04-6'], 'MeSHHeadingList': ['volkensin'], 'MeSHTermList': ['RIP protein, A volkensii Harms', 'ribosome-inactivating protein type 2, Adenia volkensii', 'volkensin'], 'PharmActionList': [], 'CommentList': [], 'IUPACName': '[(1R,2R,4R,6S,8R,11R,12S,13R,16R,17R,19S,20R)-17-acetyloxy-8-(furan-3-yl)-4,12-dihydroxy-1,9,11,16-tetramethyl-5,14-dioxapentacyclo[11.6.1.02,11.06,10.016,20]icos-9-en-19-yl] (E)-2-methylbut-2-enoate', 'CanonicalSmiles': 'CC=C(C)C(=O)OC1CC(C2(COC3C2C1(C4CC(OC5CC(C(=C5C4(C3O)C)C)C6=COC=C6)O)C)C)OC(=O)C', 'IsomericSmiles': 'C/C=C(\\C)/C(=O)O[C@H]1C[C@H]([C@]2(CO[C@@H]3[C@@H]2[C@]1([C@H]4C[C@@H](O[C@H]5C[C@H](C(=C5[C@@]4([C@@H]3O)C)C)C6=COC=C6)O)C)C)OC(=O)C', 'RotatableBondCount': IntegerElement(6, attributes={}), 'MolecularFormula': 'C33H44O9', 'MolecularWeight': '584.700', 'TotalFormalCharge': IntegerElement(0, attributes={}), 'XLogP': '3.3', 'HydrogenBondDonorCount': IntegerElement(2, attributes={}), 'HydrogenBondAcceptorCount': IntegerElement(9, attributes={}), 'Complexity': '1190.000', 'HeavyAtomCount': IntegerElement(42, attributes={}), 'AtomChiralCount': IntegerElement(12, attributes={}), 'AtomChiralDefCount': IntegerElement(12, attributes={}), 'AtomChiralUndefCount': IntegerElement(0, attributes={}), 'BondChiralCount': IntegerElement(1, attributes={}), 'BondChiralDefCount': IntegerElement(1, attributes={}), 'BondChiralUndefCount': IntegerElement(0, attributes={}), 'IsotopeAtomCount': IntegerElement(0, attributes={}), 'CovalentUnitCount': IntegerElement(1, attributes={}), 'TautomerCount': NoneElement(attributes={}), 'SubstanceIDList': [], 'TPSA': '125', 'AssaySourceNameList': [], 'MinAC': '', 'MaxAC': '', 'MinTC': '', 'MaxTC': '', 'ActiveAidCount': IntegerElement(0, attributes={}), 'InactiveAidCount': NoneElement(attributes={}), 'TotalAidCount': IntegerElement(2, attributes={}), 'InChIKey': 'KQNNSYZQMSOOQH-GLDAUDTLSA-N', 'InChI': 'InChI=1S/C33H44O9/c1-8-16(2)30(37)42-24-13-23(40-18(4)34)31(5)15-39-27-28(31)32(24,6)22-12-25(35)41-21-11-20(19-9-10-38-14-19)17(3)26(21)33(22,7)29(27)36/h8-10,14,20-25,27-29,35-36H,11-13,15H2,1-7H3/b16-8+/t20-,21+,22-,23-,24+,25-,27-,28+,29-,31-,32+,33-/m1/s1'}]
here we can see the smiles strings, this is as good as any to demonstrate the molecule module
from benchmate.molecule.molecule import Molecule
mol1=Molecule(name=mols.results[0]["Id"], smiles=mols.results[0]["CanonicalSmiles"])
mol2=Molecule(name=mols.results[1]["Id"], smiles=mols.results[1]["CanonicalSmiles"])
By default we calculate a lot of things about these molecules.
len(list(mol1.info.properties.keys()))
217
#you can compare molecules to each other
mol1.similarity(mol2, fingerprint="ecfp4")
0.8908045977011494
So far I have focused on single things, like proteins, molecules etc, I can very well be interested in genomes, let’s generate a small genome. I have manually downloaded (not shown here) a gtf file and a fasta file for the baker’s yeast genome. I’m picking yeast because its small. The generation of the databse take a little while but you have to do that once per fasta/gtf combo.
Genome, Ranges, GenomicRanges modules
These modules concern themselves as the name suggests with genomes and intervals within those genomes. I will use them togetther to show what they are capable of.
from sqlalchemy import create_engine
from benchmate.genome.genome import Genome
from benchmate.ranges.genomicranges import GenomicRange, GenomicRangesDict, GenomicRangesList
test_genome=create_engine("sqlite:///test_genome.db")
genome=Genome(name="yeast", genome_fasta="Saccharomyces_cerevisiae.R64-1-1.dna.toplevel.fa",
gtf="Saccharomyces_cerevisiae.R64-1-1.114.gtf",
description="test genome from human chrom 21 and 22",
db_conn=test_genome, standalone=True, create=False)
Found an existing genome with yeast, just setting things up, if this is an error re-initiate the class with a different name
After the genome is completes we can query for genes and other things easily.
genes=genome.genes()
len(genes.keys())
7127
You can do the same with transcripts, introns, exons, utrs and coding sequences. The syntax is pretty similar. But there are other things you can do as well. For example, you can select all the things that fall in a certain regions. For that you will need to create a genomic ranges object
myrange=GenomicRange(chrom="I", start=59, end=58654, strand="+")
interesting_genes=genome.genes(range=myrange)
interesting_genes
GenomicRangesDict(dict_items([('YAL053W', GenomicRange(I:45899-48250(+))), ('YAL064W-B', GenomicRange(I:12046-12426(+))), ('YAL056C-A', GenomicRange(I:38696-39046(-))), ('YAL048C', GenomicRange(I:52801-54789(-))), ('YAL063C-A', GenomicRange(I:22395-22685(-))), ('YAL062W', GenomicRange(I:31567-32940(+))), ('YAL045C', GenomicRange(I:57488-57796(-))), ('YAL055W', GenomicRange(I:42177-42719(+))), ('YAL060W', GenomicRange(I:35155-36303(+))), ('YAL069W', GenomicRange(I:335-649(+))), ('YAL063C', GenomicRange(I:24000-27968(-))), ('YAL046C', GenomicRange(I:57029-57385(-))), ('YAL056W', GenomicRange(I:39259-41901(+))), ('YAL066W', GenomicRange(I:10091-10399(+))), ('YAL047W-A', GenomicRange(I:54584-54913(+))), ('YAL064W', GenomicRange(I:21566-21850(+))), ('YAL054C', GenomicRange(I:42881-45022(-))), ('YAL067C', GenomicRange(I:7235-9016(-))), ('YAL059W', GenomicRange(I:36509-37147(+))), ('YAL068W-A', GenomicRange(I:538-792(+))), ('YAL065C', GenomicRange(I:11565-11951(-))), ('YAL059C-A', GenomicRange(I:36496-36918(-))), ('YAL061W', GenomicRange(I:33448-34701(+))), ('YAL067W-A', GenomicRange(I:2480-2707(+))), ('YAL049C', GenomicRange(I:51855-52595(-))), ('YAL047C', GenomicRange(I:54989-56857(-))), ('YAL044W-A', GenomicRange(I:57518-57850(+))), ('YAL068C', GenomicRange(I:1807-2169(-))), ('YAL051W', GenomicRange(I:48564-51707(+))), ('YAL044C', GenomicRange(I:57950-58462(-))), ('YAL058W', GenomicRange(I:37464-38972(+))), ('YAL064C-A', GenomicRange(I:13363-13743(-)))]))
We can see that this has returned a genomic ranges dict. This is basically a dictionary that contains genomic ranges. A genomicrange object has many features that you can check out in the documentation, you can move it around, you can see if it overlaps with another range etc.
There is another class called GenomicsRangesList, as the name suggests, it is a list of genomic ranges. Let’s create one with from the GenomicRangesDict that we have here.
gene_list=[interesting_genes[key] for key in interesting_genes.keys()]
gene_list=GenomicRangesList(gene_list)
gene_list
GenomicRangesList([GenomicRange(I:45899-48250(+)), GenomicRange(I:12046-12426(+)), GenomicRange(I:38696-39046(-)), GenomicRange(I:52801-54789(-)), GenomicRange(I:22395-22685(-)), GenomicRange(I:31567-32940(+)), GenomicRange(I:57488-57796(-)), GenomicRange(I:42177-42719(+)), GenomicRange(I:35155-36303(+)), GenomicRange(I:335-649(+)), GenomicRange(I:24000-27968(-)), GenomicRange(I:57029-57385(-)), GenomicRange(I:39259-41901(+)), GenomicRange(I:10091-10399(+)), GenomicRange(I:54584-54913(+)), GenomicRange(I:21566-21850(+)), GenomicRange(I:42881-45022(-)), GenomicRange(I:7235-9016(-)), GenomicRange(I:36509-37147(+)), GenomicRange(I:538-792(+)), GenomicRange(I:11565-11951(-)), GenomicRange(I:36496-36918(-)), GenomicRange(I:33448-34701(+)), GenomicRange(I:2480-2707(+)), GenomicRange(I:51855-52595(-)), GenomicRange(I:54989-56857(-)), GenomicRange(I:57518-57850(+)), GenomicRange(I:1807-2169(-)), GenomicRange(I:48564-51707(+)), GenomicRange(I:57950-58462(-)), GenomicRange(I:37464-38972(+)), GenomicRange(I:13363-13743(-))])
There are a few methods that makes GenomicRangesLlist worthwhile, these are overlaps and coverage. You can use another list to see the overlaps or you can calculate pariwise overlaps within itself. I will append our original ranges here to make sure that we have some overlaps.
gene_list.find_overlaps()
[(GenomicRange(I:45899-48250(+)), GenomicRange(I:45899-48250(+))),
(GenomicRange(I:12046-12426(+)), GenomicRange(I:12046-12426(+))),
(GenomicRange(I:38696-39046(-)), GenomicRange(I:38696-39046(-))),
(GenomicRange(I:52801-54789(-)), GenomicRange(I:52801-54789(-))),
(GenomicRange(I:22395-22685(-)), GenomicRange(I:22395-22685(-))),
(GenomicRange(I:31567-32940(+)), GenomicRange(I:31567-32940(+))),
(GenomicRange(I:57488-57796(-)), GenomicRange(I:57488-57796(-))),
(GenomicRange(I:42177-42719(+)), GenomicRange(I:42177-42719(+))),
(GenomicRange(I:35155-36303(+)), GenomicRange(I:35155-36303(+))),
(GenomicRange(I:335-649(+)), GenomicRange(I:335-649(+))),
(GenomicRange(I:24000-27968(-)), GenomicRange(I:24000-27968(-))),
(GenomicRange(I:57029-57385(-)), GenomicRange(I:57029-57385(-))),
(GenomicRange(I:39259-41901(+)), GenomicRange(I:39259-41901(+))),
(GenomicRange(I:10091-10399(+)), GenomicRange(I:10091-10399(+))),
(GenomicRange(I:54584-54913(+)), GenomicRange(I:54584-54913(+))),
(GenomicRange(I:21566-21850(+)), GenomicRange(I:21566-21850(+))),
(GenomicRange(I:42881-45022(-)), GenomicRange(I:42881-45022(-))),
(GenomicRange(I:7235-9016(-)), GenomicRange(I:7235-9016(-))),
(GenomicRange(I:36509-37147(+)), GenomicRange(I:36509-37147(+))),
(GenomicRange(I:538-792(+)), GenomicRange(I:538-792(+))),
(GenomicRange(I:11565-11951(-)), GenomicRange(I:11565-11951(-))),
(GenomicRange(I:36496-36918(-)), GenomicRange(I:36496-36918(-))),
(GenomicRange(I:33448-34701(+)), GenomicRange(I:33448-34701(+))),
(GenomicRange(I:2480-2707(+)), GenomicRange(I:2480-2707(+))),
(GenomicRange(I:51855-52595(-)), GenomicRange(I:51855-52595(-))),
(GenomicRange(I:54989-56857(-)), GenomicRange(I:54989-56857(-))),
(GenomicRange(I:57518-57850(+)), GenomicRange(I:57518-57850(+))),
(GenomicRange(I:1807-2169(-)), GenomicRange(I:1807-2169(-))),
(GenomicRange(I:48564-51707(+)), GenomicRange(I:48564-51707(+))),
(GenomicRange(I:57950-58462(-)), GenomicRange(I:57950-58462(-))),
(GenomicRange(I:37464-38972(+)), GenomicRange(I:37464-38972(+))),
(GenomicRange(I:13363-13743(-)), GenomicRange(I:13363-13743(-)))]
This is kind of a silly example because the only overlaps here are with self, but you get the idea. Each of these genomic ranges, contains some sort of annotation that comes with the gtf file, these depend on where you get your annotations. This one is from ensembl and it lookst like this:
gene_list[0].annotation
{'gene_id': 'YAL053W',
'gene_name': 'FLC2',
'gene_source': 'sgd',
'gene_biotype': 'protein_coding',
'db_id': 6950}
You can use these annotations to filter your results (like get all protein coding genes) or even better you ca use them to search other things.
uniprot.search(gene_list[0].annotation["gene_id"])
[{'id': 'P39719',
'gene': 'FLC2',
'description': ['Flavin carrier protein 2',
['FAD transporter 2', 'TRP-like ion channel FLC2']],
'synonyms': [],
'organism': 'Saccharomyces cerevisiae (strain ATCC 204508 / S288c)'},
{'id': 'J8Q898',
'gene': None,
'description': ['YAL053W'],
'synonyms': [],
'organism': 'Saccharomyces arboricola (strain H-6 / AS 2.3317 / CBS 10644)'},
{'id': 'J4TTV2',
'gene': 'YAL053W',
'description': ['FLC2-like protein'],
'synonyms': ['SKDI01G0150'],
'organism': 'Saccharomyces kudriavzevii (strain ATCC MYA-4449 / AS 2.2408 / CBS 8840 / NBRC 1802 / NCYC 2889)'},
{'id': 'A0A0J9XGZ3',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter'],
'synonyms': ['SKDI01G0150'],
'organism': 'Geotrichum candidum'},
{'id': 'A0A0J9XFH0',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter'],
'synonyms': ['SKDI01G0150'],
'organism': 'Geotrichum candidum'},
{'id': 'A0A0J9XHC4',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter required for uptake of FAD into endoplasmic reticulum'],
'synonyms': ['SKDI01G0150'],
'organism': 'Geotrichum candidum'},
{'id': 'A0A8H2ZME8',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter'],
'synonyms': ['SKDI01G0150'],
'organism': 'Maudiozyma barnettii'},
{'id': 'A0A1X7QZG9',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter'],
'synonyms': ['SKDI01G0150'],
'organism': 'Maudiozyma saulgeensis'},
{'id': 'A0A0J9XJC6',
'gene': None,
'description': ['Similar to Saccharomyces cerevisiae YAL053W FLC2 Putative FAD transporter, required for uptake of FAD into endoplasmic reticulum'],
'synonyms': ['SKDI01G0150'],
'organism': 'Geotrichum candidum'}]
Last but not least on the genome module you can get arbitrary sequences for a given range.
genome.get_sequence(gene_list[0])
'TGATCTTCCTAAACACCTTCGCAAGGTGCCTTTTAACGTGTTTCGTACTGTGCAGCGGTACAGCACGTTCCTCTGACACAAACGACACTACTCCGGCGTCTGCAAAGCATTTGCAGACCACTTCTTTATTGACGTGTATGGACAATTCGCAATTAACGGCATCATTCTTTGATGTGAAATTTTACCCCGATAATAATACTGTTATCTTTGATATTGACGCTACGACGACGCTTAATGGGAACGTCACTGTGAAGGCTGAGCTGCTTACTTACGGACTGAAAGTCCTGGATAAGACTTTTGATTTATGTTCCTTGGGCCAAGTATCGCTTTGCCCCCTAAGTGCTGGGCGTATTGATGTCATGTCCACACAGGTGATCGAATCATCCATTACCAAGCAATTTCCCGGCATTGCTTACACCATTCCAGATTTGGACGCACAAGTACGTGTGGTGGCATACGCTCAGAATGACACGGAATTCGAAACTCCGCTGGCTTGTGTCCAGGCTATCTTGAGTAACGGGAAGACAGTGCAAACAAAGTATGCGGCCTGGCCCATTGCCGCTATCTCAGGTGTCGGTGTACTTACCTCAGGGTTTGTGTCTGTGATCGGTTACTCAGCCACTGCTGCTCACATTGCGTCCAACTCCATCTCATTGTTCATATACTTCCAAAATCTAGCTATCACTGCAATGATGGGTGTCTCAAGGGTTCCACCCATTGCTGCCGCGTGGACGCAGAATTTCCAATGGTCCATGGGTATCATCAATACAAACTTCATGCAAAAGATTTTTGATTGGTACGTACAGGCCACTAATGGTGTCTCAAATGTTGTGGTAGCTAACAAGGACGTCTTGTCCATTAGTGTGCAAAAACGTGCTATCTCTATGGCATCGTCTAGTGATTACAATTTTGACACCATTTTAGACGATTCGAATCTGTACACCACTTCTGAGAAGGATCCAAGCAATTACTCAGCCAAGATTCTCGTGTTAAGAGGTATAGAAAGAGTTGCTTATTTGGCTAATATTGAGCTATCTAATTTCTTTTTGACCGGTATTGTGTTTTTTCTATTCTTCCTATTTGTAGTTGTCGTCTCTTTGATTTTCTTTAAGGCGCTATTGGAAGTTCTTACAAGAGCAAGAATATTGAAAGAGACTTCCAATTTCTTCCAATATAGGAAGAACTGGGGGAGTATTATCAAAGGCACCCTTTTCAGATTATCTATCATCGCCTTCCCTCAAGTTTCTCTTCTGGCGATTTGGGAATTTACTCAGGTCAACTCTCCAGCGATTGTTGTTGATGCGGTAGTAATATTACTGATCATCACGGGACTTCTGGTTTATGGAACTATAAGGGTTTTCATCAAGGGAAGAGAGTCTCTCAGATTATACAAGAATCCTGCGTACCTACTTTACAGTGATACCTACTTCTTGAACAAGTTTGGGTTCTTATACGTTCAATTCAAAGCAGATAAGTTTTGGTGGCTTTTACCCTTATTAAGTTATGCGTTCTTAAGATCCCTGTTTGTTGCCGTTTTACAAAACCAAGGTAAGGCTCAAGCAATGATCATCTTTGTCATTGAACTAGCTTACTTCGTTTGTCTCTGTTGGATAAGACCATATTTGGACAAGAGAACTAATGTTTTCAATATTGCTATTCATTTGGTGAATTTGATCAATGCATTTTTCTTTTTGTTTTTCAGTAATTTGTTCAAGCAACCAGCAGTGGTTTCGTCAGTGATGGCGGTTATTCTGTTCGTTTTGAACGCGGTGTTTGCTCTATTCCTATTATTGTTCACTATTGTCACCTGTACACTGGCATTACTACACAGAAACCCAGATGTCCGTTACCAACCAATGAAAGATGACCGTGTGTCATTCATTCCTAAGATTCAAAATGATTTCGATGGCAAAAACAAAAATGATTCTGAACTGTTTGAATTGAGAAAAGCTGTTATGGACACCAATGAAAATGAGGAAGAAAAAATGTTCCGTGACGACACTTTCGGCAAGAACCTGAATGCAAACACAAATACAGCAAGACTCTTTGATGATGAGACTAGTTCATCCTCTTTTAAGCAAAATTCCTCTCCCTTCGATGCCTCGGAAGTAACGGAGCAACCTGTGCAACCAACCTCCGCTGTCATGGGTACGGGTGGCAGCTTCTTGTCTCCACAGTACCAACGTGCGTCATCTGCTTCTCGTACTAATCTAGCGCCGAATAATACAAGCACCTCCAGTTTAATGAAGCCTGAATCAAGTCTCTACCTGGGGAATTCCAATAAATCATATTCGCATTTTAACAACAACGGCAGCAACGAAAACGCCCGCAACAACAACCCATATTTGTAA'
Alignment module
Speaking of sequences, we love comparing sequences to each other, because we love them so much, we’ve written a whole module for it. The alignment module allows you to perform local searches using blast, mmseqs2 and foldseek. You can see what they are in the links. Here I will quickly demonstrate how to use mmseqs, this is helpful if you are interested in evolutionary conservation or just trying to genearate an MSA for other purposes like feeding into AF3.
from benchmate.alignment.mmseqs import MMSeqs
mmseqs=MMSeqs()
mmseqs.list_dbs()
['usage: mmseqs databases <name> <o:sequenceDB> <tmpDir> [options]',
'',
' Name \tType \tTaxonomy\tUrl ',
'- UniRef100 \tAminoacid \t yes\thttps://www.uniprot.org/help/uniref',
'- UniRef90 \tAminoacid \t yes\thttps://www.uniprot.org/help/uniref',
'- UniRef50 \tAminoacid \t yes\thttps://www.uniprot.org/help/uniref',
'- UniProtKB \tAminoacid \t yes\thttps://www.uniprot.org/help/uniprotkb',
'- UniProtKB/TrEMBL \tAminoacid \t yes\thttps://www.uniprot.org/help/uniprotkb',
'- UniProtKB/Swiss-Prot\tAminoacid \t yes\thttps://uniprot.org',
'- NR \tAminoacid \t yes\thttps://ftp.ncbi.nlm.nih.gov/blast/db/FASTA',
'- NT \tNucleotide\t -\thttps://ftp.ncbi.nlm.nih.gov/blast/db/FASTA',
'- GTDB \tAminoacid \t yes\thttps://gtdb.ecogenomic.org',
'- PDB \tAminoacid \t -\thttps://www.rcsb.org',
'- PDB70 \tProfile \t -\thttps://github.com/soedinglab/hh-suite',
'- Pfam-A.full \tProfile \t -\thttps://pfam.xfam.org',
'- Pfam-A.seed \tProfile \t -\thttps://pfam.xfam.org',
'- Pfam-B \tProfile \t -\thttps://xfam.wordpress.com/2020/06/30/a-new-pfam-b-is-released',
'- CDD \tProfile \t -\thttps://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml',
'- eggNOG \tProfile \t -\thttp://eggnog5.embl.de',
'- VOGDB \tProfile \t -\thttps://vogdb.org',
'- dbCAN2 \tProfile \t -\thttp://bcb.unl.edu/dbCAN2',
'- SILVA \tNucleotide\t yes\thttps://www.arb-silva.de',
'- Resfinder \tNucleotide\t -\thttps://cge.cbs.dtu.dk/services/ResFinder',
'- Kalamari \tNucleotide\t yes\thttps://github.com/lskatz/Kalamari',
'options: ',
' --tsv BOOL Return output in TSV format [0]',
' ',
' --compressed INT Write compressed output [0]',
' --threads INT Number of CPU-cores used (all by default) [20]',
' -v INT Verbosity level: 0: quiet, 1: +errors, 2: +warnings, 3: +info [3]',
'',
'references:',
' - Steinegger M, Soding J: MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nature Biotechnology, 35(11), 1026-1028 (2017)',
' - Mirdita M, Steinegger M, Breitwieser F, Soding J, Levy Karin E: Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics, btab184 (2021)',
'',
"Show an extended list of options by calling 'mmseqs databases -h'."]
These are all the available databases that you can download. Let’s download swissprot
mmseqs.download_db("UniProtKB/Swiss-Prot", "./mmseqs", create=True)
databases UniProtKB/Swiss-Prot ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmpeugtd5fc
MMseqs Version: 18.8cc5c
Tsv false
Force restart with latest tmp false
Remove temporary files false
Compressed 0
Threads 20
Verbosity 3
11/06 11:44:51 [[1;32mNOTICE[0m] Downloading 1 item(s)
11/06 11:44:51 [[1;32mNOTICE[0m] Download complete: /tmp/tmpeugtd5fc/15116153508622793925/version.aria2
Download Results:
gid |stat|avg speed |path/URI
======+====+===========+=======================================================
2acc25|OK | 18KiB/s|/tmp/tmpeugtd5fc/15116153508622793925/version.aria2
Status Legend:
(OK):download completed.
11/06 11:44:51 [[1;32mNOTICE[0m] Downloading 1 item(s)
[#7a297f 1.0MiB/88MiB(1%) CN:4 DL:1.6MiB ETA:52s]
[#7a297f 34MiB/88MiB(39%) CN:4 DL:21MiB ETA:2s]
[#7a297f 84MiB/88MiB(94%) CN:3 DL:32MiB]
11/06 11:44:55 [[1;32mNOTICE[0m] Download complete: /tmp/tmpeugtd5fc/15116153508622793925/uniprot_sprot.fasta.gz.aria2
Download Results:
gid |stat|avg speed |path/URI
======+====+===========+=======================================================
7a297f|OK | 31MiB/s|/tmp/tmpeugtd5fc/15116153508622793925/uniprot_sprot.fasta.gz.aria2
Status Legend:
(OK):download completed.
createdb /tmp/tmpeugtd5fc/15116153508622793925/uniprot_sprot.fasta.gz ./mmseqs/UniProtKB_Swiss-Prot --compressed 0 -v 3 --gpu 1
Converting sequences
[=========================================================
Sort single files in 0h 0m 0s 409ms
Merge all files 0h 0m 0s 418ms
Database type: Aminoacid
Time for processing: 0h 0m 3s 375ms
prefixid ./mmseqs/UniProtKB_Swiss-Prot_h /tmp/tmpeugtd5fc/15116153508622793925/header_pref.tsv --tsv --threads 20 -v 3
[=================================================================] 573.66K 0s 57ms
Time for merging to header_pref.tsv: 0h 0m 0s 151ms
Time for processing: 0h 0m 0s 282ms
Create directory /tmp/tmpeugtd5fc/15116153508622793925/taxonomy
createtaxdb ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmpeugtd5fc/15116153508622793925/taxonomy --threads 20 -v 3
Download taxdump.tar.gz
11/06 11:45:00 [[1;32mNOTICE[0m] Downloading 1 item(s)
[#31da40 11MiB/67MiB(16%) CN:2 DL:13MiB ETA:4s]
[#31da40 42MiB/67MiB(63%) CN:2 DL:23MiB ETA:1s]
11/06 11:45:03 [[1;32mNOTICE[0m] Download complete: /tmp/tmpeugtd5fc/15116153508622793925/taxonomy/taxdump.tar.gz.aria2
Download Results:
gid |stat|avg speed |path/URI
======+====+===========+=======================================================
31da40|OK | 24MiB/s|/tmp/tmpeugtd5fc/15116153508622793925/taxonomy/taxdump.tar.gz.aria2
Status Legend:
(OK):download completed.
Loading nodes file ... Done, got 2705917 nodes
Loading merged file ... Done, added 93443 merged nodes.
Loading names file ... Done
Init computeSparseTable ...Done
'./mmseqs/UniProtKB_Swiss-Prot'
Ok, our databsae is ready and the name we are going to use is returned with the function. Remember the sequence instance we created way back when with upf1? Let’s use that one for alignment.
results=mmseqs.search(myseq, "./mmseqs/UniProtKB_Swiss-Prot", output_tsv="upf1.tsv", output_a3m="upf1.a3m")
createdb /tmp/tmp6q1ifu8x/query.fasta /tmp/tmp6q1ifu8x/query_db
MMseqs Version: 18.8cc5c
Database type 0
Shuffle input database true
Createdb mode 0
Write lookup file 1
Offset of numeric ids 0
Threads 20
Compressed 0
Mask residues 0
Mask residues probability 0.9
Mask lower case residues 0
Mask lower letter repeating N times 0
Use GPU 0
Verbosity 3
Converting sequences
[
Time for merging to query_db_h: 0h 0m 0s 0ms
Time for merging to query_db: 0h 0m 0s 0ms
Database type: Aminoacid
Time for processing: 0h 0m 0s 6ms
search /tmp/tmp6q1ifu8x/query_db ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmp6q1ifu8x/result /tmp/tmp6q1ifu8x --max-seqs 1000 -s 5.7 -e 0.001
MMseqs Version: 18.8cc5c
Substitution matrix aa:blosum62.out,nucl:nucleotide.out
Add backtrace false
Alignment mode 2
Alignment mode 0
Allow wrapped scoring false
E-value threshold 0.001
Seq. id. threshold 0
Min alignment length 0
Seq. id. mode 0
Alternative alignments 0
Coverage threshold 0
Coverage mode 0
Max sequence length 65535
Compositional bias 1
Compositional bias scale 1
Max reject 2147483647
Max accept 2147483647
Include identical seq. id. false
Preload mode 0
Pseudo count a substitution:1.100,context:1.400
Pseudo count b substitution:4.100,context:5.800
Score bias 0
Realign hits false
Realign score bias -0.2
Realign max seqs 2147483647
Correlation score weight 0
Gap open cost aa:11,nucl:5
Gap extension cost aa:1,nucl:2
Zdrop 40
Threads 20
Compressed 0
Verbosity 3
Seed substitution matrix aa:VTML80.out,nucl:nucleotide.out
Sensitivity 5.7
k-mer length 0
Target search mode 0
k-score seq:2147483647,prof:2147483647
Alphabet size aa:21,nucl:5
Max results per query 1000
Split database 0
Split mode 2
Split memory limit 0
Diagonal scoring true
Exact k-mer matching 0
Mask residues 1
Mask residues probability 0.9
Mask lower case residues 0
Mask lower letter repeating N times 0
Minimum diagonal score 15
Selected taxa
Spaced k-mers 1
Spaced k-mer pattern
Local temporary path
Use GPU 0
Use GPU server 0
Wait for GPU server 600
Prefilter mode 0
Rescore mode 0
Remove hits by seq. id. and coverage false
Sort results 0
Mask profile 1
Profile E-value threshold 0.1
Global sequence weighting false
Allow deletions false
Filter MSA 1
Use filter only at N seqs 0
Maximum seq. id. threshold 0.9
Minimum seq. id. 0.0
Minimum score per column -20
Minimum coverage 0
Select N most diverse seqs 1000
Pseudo count mode 0
Profile output mode 0
Min codons in orf 30
Max codons in length 32734
Max orf gaps 2147483647
Contig start mode 2
Contig end mode 2
Orf start mode 1
Forward frames 1,2,3
Reverse frames 1,2,3
Translation table 1
Translate orf 0
Use all table starts false
Offset of numeric ids 0
Create lookup 0
Overlap between sequences 0
Sequence split mode 1
Header split mode 0
Chain overlapping alignments 0
Merge query 1
Search type 0
Search iterations 1
Start sensitivity 4
Search steps 1
Exhaustive search mode false
Filter results during exhaustive search 0
Strand selection 1
LCA search mode false
Disk space limit 0
MPI runner
Force restart with latest tmp false
Remove temporary files false
Translation mode 0
prefilter /tmp/tmp6q1ifu8x/query_db ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmp6q1ifu8x/16443233877141691215/pref_0 --sub-mat 'aa:blosum62.out,nucl:nucleotide.out' --seed-sub-mat 'aa:VTML80.out,nucl:nucleotide.out' -k 0 --target-search-mode 0 --k-score seq:2147483647,prof:2147483647 --alph-size aa:21,nucl:5 --max-seq-len 65535 --max-seqs 1000 --split 0 --split-mode 2 --split-memory-limit 0 -c 0 --cov-mode 0 --comp-bias-corr 1 --comp-bias-corr-scale 1 --diag-score 1 --exact-kmer-matching 0 --mask 1 --mask-prob 0.9 --mask-lower-case 0 --mask-n-repeat 0 --min-ungapped-score 15 --add-self-matches 0 --spaced-kmer-mode 1 --db-load-mode 0 --pca substitution:1.100,context:1.400 --pcb substitution:4.100,context:5.800 --threads 20 --compressed 0 -v 3 -s 5.7
Query database size: 1 type: Aminoacid
Estimated memory consumption: 2G
Target database size: 573661 type: Aminoacid
Index table k-mer threshold: 112 at k-mer size 6
Index table: counting k-mers
[=================================================================] 573.66K 2s 11ms
Index table: Masked residues: 3280229
Index table: fill
[=================================================================] 573.66K 2s 204ms
Index statistics
Entries: 197795838
DB size: 1620 MB
Avg k-mer size: 3.090560
Top 10 k-mers
GPGGTL 1852
GQSWTV 1705
MSWAIT 1697
GYLWMS 1666
ILSPLG 1662
AFGVWG 1650
YVPGSF 1637
WGMFAT 1637
PGVFEV 1637
VLWQFW 1622
Time for index table init: 0h 0m 4s 778ms
Process prefiltering step 1 of 1
k-mer similarity threshold: 112
Starting prefiltering scores calculation (step 1 of 1)
Query db start 1 to 1
Target db start 1 to 573661
[=================================================================] 1 0s 0ms
359.934455 k-mers per position
1049853 DB matches per sequence
0 overflows
1000 sequences passed prefiltering per query sequence
1000 median result list length
0 sequences with 0 size result lists
Time for merging to pref_0: 0h 0m 0s 0ms
Time for processing: 0h 0m 5s 52ms
align /tmp/tmp6q1ifu8x/query_db ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmp6q1ifu8x/16443233877141691215/pref_0 /tmp/tmp6q1ifu8x/result --sub-mat 'aa:blosum62.out,nucl:nucleotide.out' -a 0 --alignment-mode 2 --alignment-output-mode 0 --wrapped-scoring 0 -e 0.001 --min-seq-id 0 --min-aln-len 0 --seq-id-mode 0 --alt-ali 0 -c 0 --cov-mode 0 --max-seq-len 65535 --comp-bias-corr 1 --comp-bias-corr-scale 1 --max-rejected 2147483647 --max-accept 2147483647 --add-self-matches 0 --db-load-mode 0 --pca substitution:1.100,context:1.400 --pcb substitution:4.100,context:5.800 --score-bias 0 --realign 0 --realign-score-bias -0.2 --realign-max-seqs 2147483647 --corr-score-weight 0 --gap-open aa:11,nucl:5 --gap-extend aa:1,nucl:2 --zdrop 40 --threads 20 --compressed 0 -v 3
Compute score and coverage
Query database size: 1 type: Aminoacid
Target database size: 573661 type: Aminoacid
Calculation of alignments
[=================================================================] 1 0s 0ms
Time for merging to result: 0h 0m 0s 0ms
1000 alignments calculated
72 sequence pairs passed the thresholds (0.072000 of overall calculated)
72.000000 hits per query sequence
Time for processing: 0h 0m 0s 107ms
result2msa /tmp/tmp6q1ifu8x/query_db ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmp6q1ifu8x/result /tmp/tmp6q1ifu8x/result.a3m
MMseqs Version: 18.8cc5c
Substitution matrix aa:blosum62.out,nucl:nucleotide.out
Gap open cost aa:11,nucl:5
Gap extension cost aa:1,nucl:2
Allow deletions false
Compositional bias 1
Compositional bias scale 1
MSA format mode 2
Summary prefix cl
Skip query false
Filter MSA 0
Use filter only at N seqs 0
Maximum seq. id. threshold 0.9
Minimum seq. id. 0.0
Minimum score per column -20
Minimum coverage 0
Select N most diverse seqs 1000
Preload mode 0
Threads 20
Compressed 0
Verbosity 3
Query database size: 1 type: Aminoacid
Target database size: 573661 type: Aminoacid
[=================================================================] 1 0s 1ms
Time for merging to result.a3m: 0h 0m 0s 0ms
Time for processing: 0h 0m 0s 111ms
convertalis /tmp/tmp6q1ifu8x/query_db ./mmseqs/UniProtKB_Swiss-Prot /tmp/tmp6q1ifu8x/result upf1.tsv --format-output query,target,pident,alnlen,mismatch,gapopen,qstart,qend,tstart,tend,evalue,bits
MMseqs Version: 18.8cc5c
Substitution matrix aa:blosum62.out,nucl:nucleotide.out
Alignment format 0
Format alignment output query,target,pident,alnlen,mismatch,gapopen,qstart,qend,tstart,tend,evalue,bits
Translation table 1
Gap open cost aa:11,nucl:5
Gap extension cost aa:1,nucl:2
Database output false
Preload mode 0
Search type 0
Threads 20
Compressed 0
Verbosity 3
[=================================================================] 1 0s 0ms
Time for merging to upf1.tsv: 0h 0m 0s 0ms
Time for processing: 0h 0m 0s 50ms
results
('upf1.a3m', 'upf1.tsv')
Here are the files that we got after alignment.
Conclusions
As we can see there are many ways we can use the individual modules withing the package to intearct with each other. There are many many features of benchmate that we have not touched. Please take a look at the detailed documentation for all the methods that are available in each module.
If you run into problems, please create an issue in the git repository here